【科学背景】
蛋白质并非静态的刚性分子,而是在其低能天然态与多种高能构象(如部分或完全去折叠状态)之间持续动态转换。这些高能态尽管在热力学上占比极小,却在蛋白质功能、相互作用、聚集行为及免疫原性等过程中发挥关键调控作用。然而,受限于传统结构生物学方法的局限,这类构象波动长期被视为“不可见”的,其能量特性与序列决定规律至今知之甚少。尽管以AlphaFold为代表的AI技术已能高精度预测蛋白质天然结构,但它们在预测折叠稳定性或构象能量差异方面依然力不从心。当前亟需一种高通量实验手段,能系统解析序列空间内蛋白质能量景观的多样性,从而为物理模型与机器学习方法的迭代提供数据支撑。

【创新成果】
近日,美国西北大学范伯格医学院Gabriel J. Rocklin团队等研究者在Nature上发表了题为“Large-scale discovery, analysis and design of protein energy landscapes”的论文,报道了一种多重氢氘交换质谱(mHDX-MS)策略,首次实现了对数百个蛋白质结构域构象波动能量的大规模并行分析。通过对5,778个长度在28-64个氨基酸残基之间的天然与人工设计结构域进行测定,研究揭示了不同序列间能量景观的显著差异,这类差异即便在具有相同折叠拓扑和相似全局稳定性的蛋白质中依然存在。基于实验数据,作者定义了“开放协同性”这一指标,用以量化蛋白质在去折叠过程中局部结构单元的能量异质性。进一步结合定点氢氘交换核磁共振与结构建模,发现低协同性的结构域往往存在特定二级结构元件的局部失稳。通过系统分析序列-结构特征与协同性的关联,并利用机器学习模型指导突变设计,作者成功筛选出能够增强局部稳定性并改善协同性的双点突变体。该研究不仅提供了迄今规模最大的蛋白质能量景观实验数据集,还展示了mHDX-MS技术在蛋白质工程与基础生物物理研究中的广阔应用前景。
【数据概览】
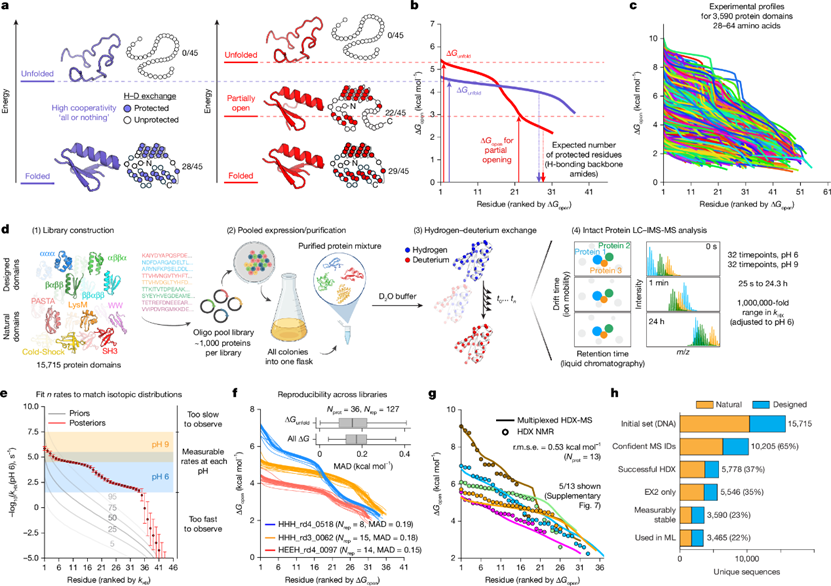
图1 mHDX-MS方法用于绘制蛋白质能量景观的基本原理与工作流程©2026 Nature
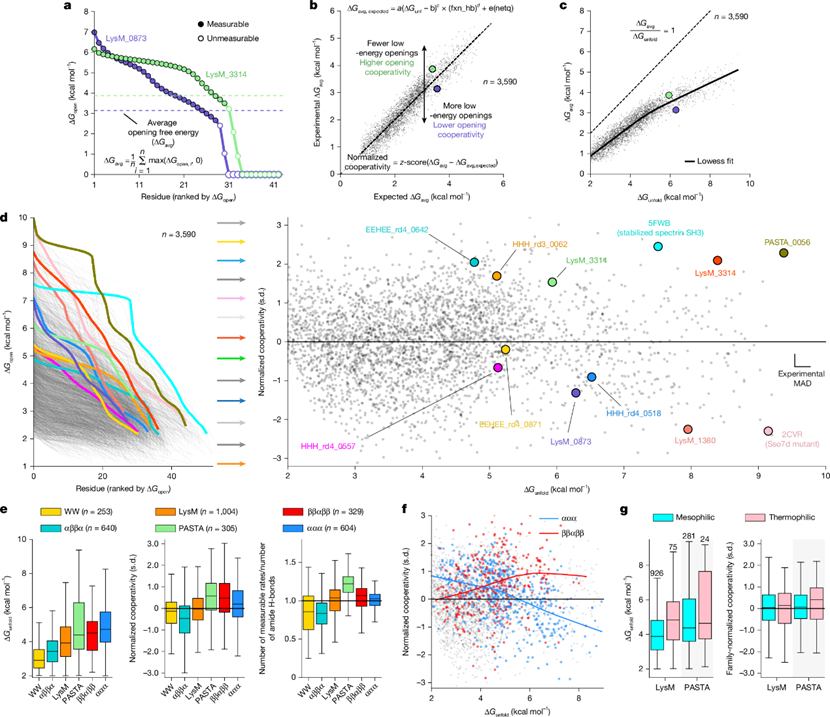
图2 小结构域的稳定性与开放协同性差异©2026 Nature
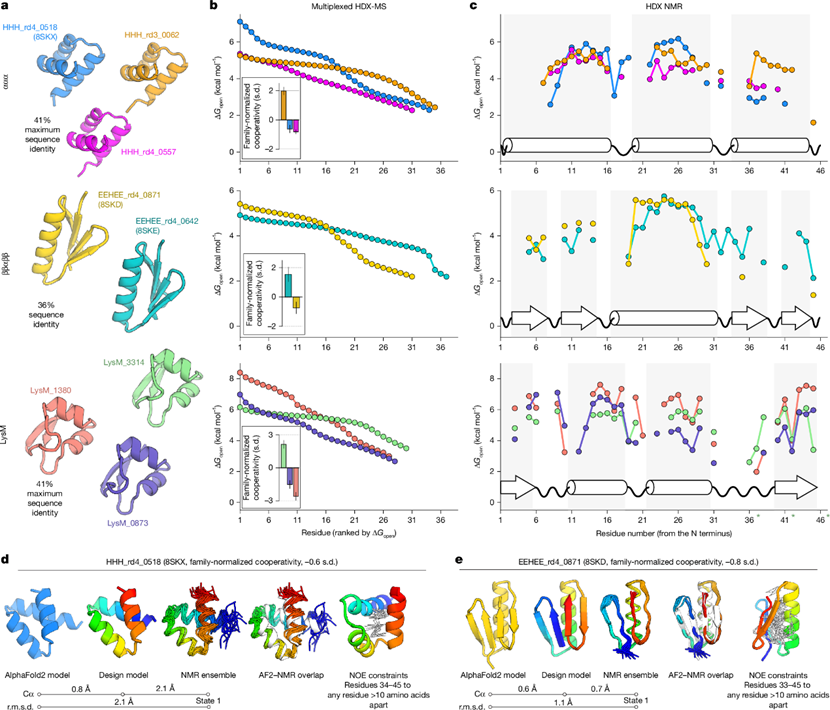
图3 低协同性结构域的不稳定残基呈簇集分布©2026 Nature
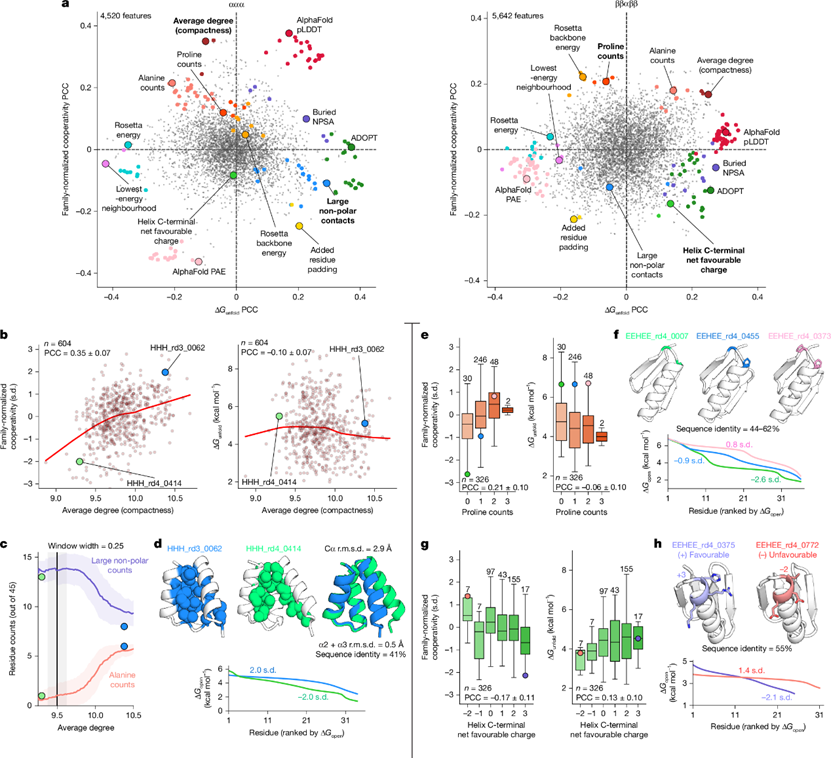
图4 与开放协同性相关的蛋白质特征©2026 Nature
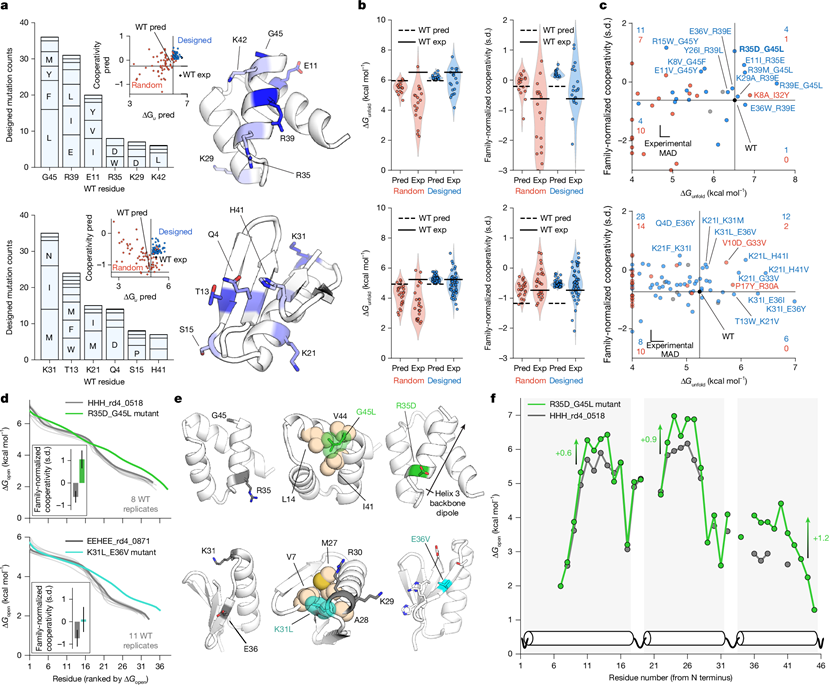
图5 数据驱动的开放协同性优化设计©2026 Nature
【科学启迪】
该项研究证实,蛋白质构象波动在不同序列间存在远超预期的多样性,且这种多样性在传统结构预测中完全无法体现。通过mHDX-MS技术,研究得以在千级别规模上量化开放协同性,发现低协同性通常源于特定二级结构元件的局部能量劣势。尽管全球折叠稳定性与局部残基稳定性在部分结构域中呈现解耦趋势,二者之间的关联难以用单一序列特征准确描述。机器学习模型虽能捕捉部分趋势,但对协同性的预测能力仍远逊于对全局稳定性的预测(R²仅0.16–0.24),表明当前特征集尚无法完整描述能量景观的调控机制。未来,通过结合更高分辨率的质谱碎片技术与生成式AI模型,mHDX-MS有望成为解析蛋白质非天然态空间的关键工具,推动新药靶点发现、免疫原性预测及稳定性工程化改造的快速发展。
原文链接:https://doi.org/10.1038/s41586-026-10465-z