一、 【研究背景】
天然酶是维持生命活动的关键,但其高成本、低稳定性和保存困难限制了其在医疗与工业中的广泛应用。近年来,单原子催化剂(SACs)因具备最大化的原子利用率和天然金属酶相似的原子级催化位点,成为新一代人工酶的研究热点。然而,SACs通常仅含单一类型的活性位点,难以突破多步酶催化反应中的线性比例关系限制。例如,增强底物吸附能力的同时往往会抑制产物的脱附,反之亦然。为突破该瓶颈,构建双原子催化剂(DACs)成为一种颇具前景的策略。尽管DACs展现出巨大潜力,其研究仍处于起步阶段,普遍存在结构界定不明确和构效关系不清晰等问题。目前许多所谓的DACs中,真正参与催化反应的往往仍是单一金属原子,另一原子仅起到调控电子结构的作用。此外,引入非金属原子作为第二催化位点的潜力尚未被充分挖掘。而在自然界中,许多金属酶之所以具有卓越的催化能力,正是源自中心金属原子与邻近氨基酸残基中非金属原子的协同作用。
二、【成果掠影】
受天然金属酶启发,新加坡南洋理工大学陈鹏教授、香港城市大学刘彬教授、山西医科大学李利平教授等研究人员报道了一种仿生催化对,将具有强电子耦合的锰–硫原子对锚定在氮硫共掺杂空心碳球上(Mn1‒S/NSHCS),用于协同驱动多种类酶催化反应。该催化对中,非金属硫原子不仅能够调控金属锰位点的电子结构,增强底物的吸附和活化能力,同时作为第二催化位点参与反应中间体的稳定和产物脱附,有效打破了传统SACs中吸附与脱附难以兼顾的瓶颈。相关研究以”Atomic Metal–Nonmetal Catalytic Pair Cooperatively Drives Efficient Enzyme-Mimetic Catalysis”为题,发表在《Angewandte Chemie International Edition》期刊上。

三、【数据概览】
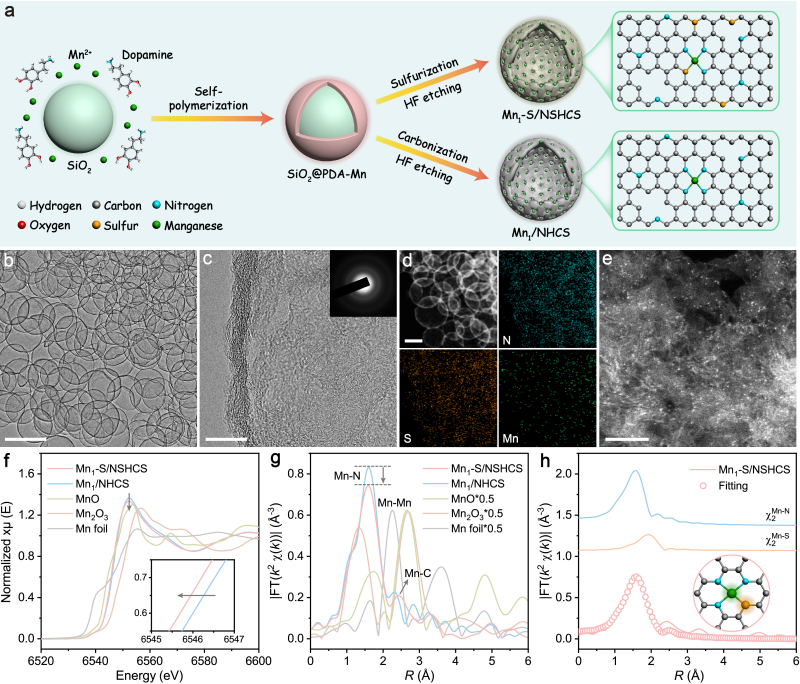
图1. Mn1–S/NSHCS的设计合成和结构表征
Mn1‒S/NSHCS催化剂通过模板辅助的高温硫化策略合成。结构表征结果显示,该催化剂呈现规则的中空球壳形貌,锰原子以单原子形式均匀分布于碳球表面,并与氮、硫原子配位形成不对称的Mn–N3S配位结构。
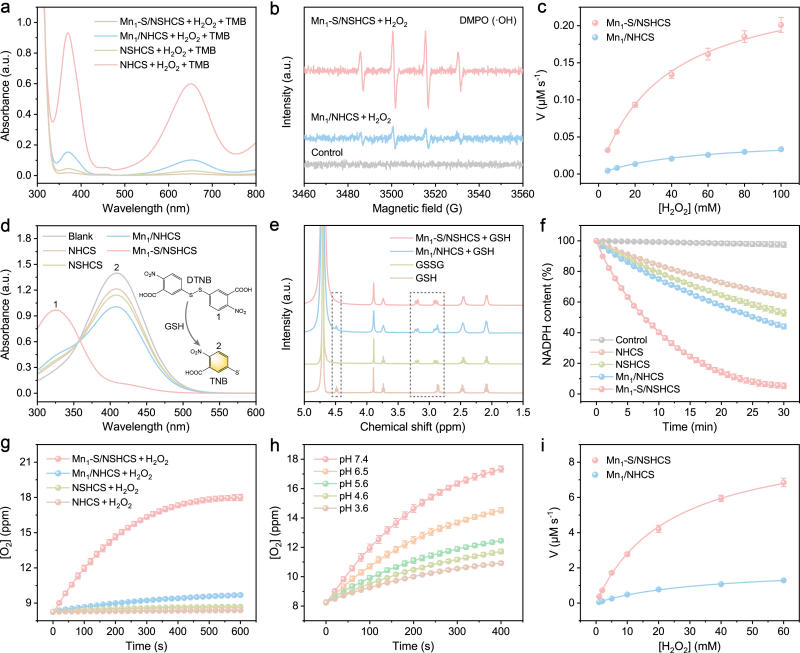
图2. 类酶催化活性评估
Mn1‒S/NSHCS展现出优异的类多酶活性,包括类过氧化物酶(POD),谷胱甘肽氧化酶(GSHOx),NADPH氧化酶(NOX)和过氧化氢酶(CAT)。
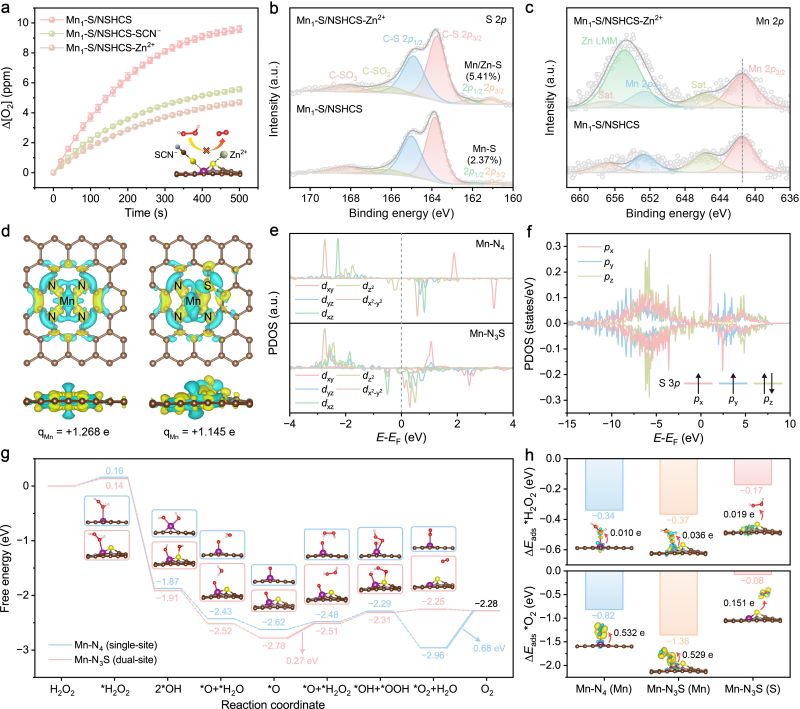
图3. 催化机制研究
催化位点毒化实验和理论计算表明,Mn–S催化对的协同可同时促进底物H2O2在Mn位点上的吸附与活化,以及产物O2在S位点上的快速脱附,打破了传统SACs中吸附与脱附难以兼顾的限制,从而增强类酶活性。
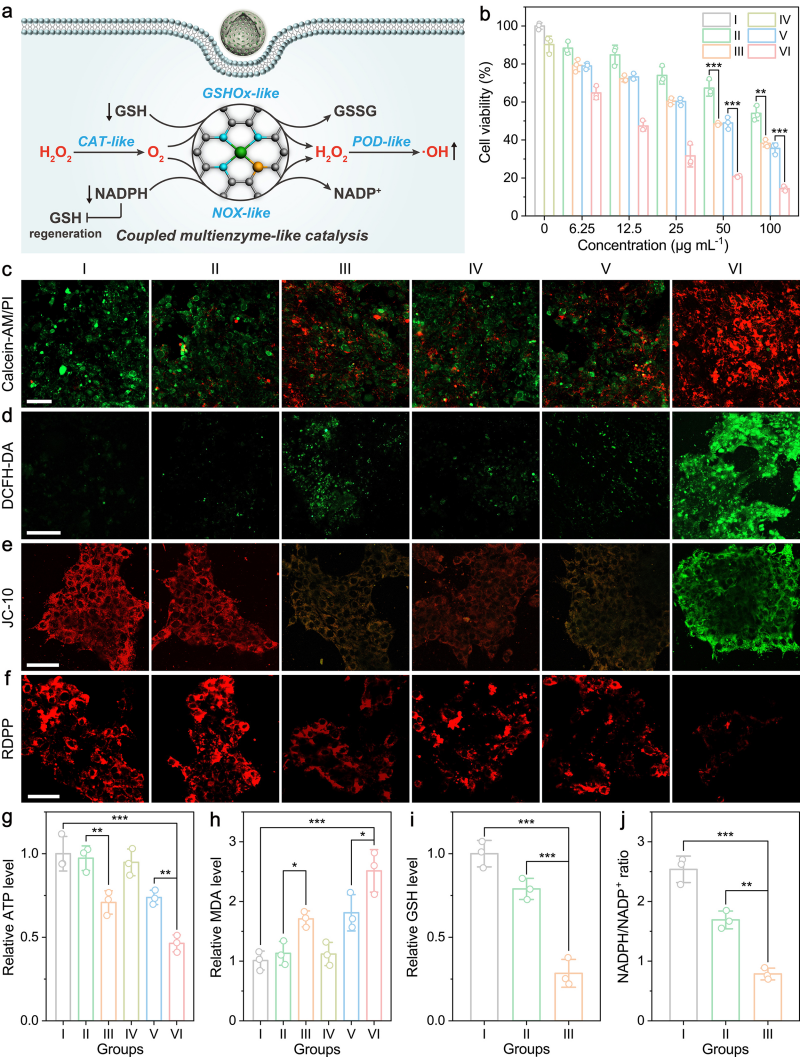
图4. 体外治疗效果
Mn1‒S/NSHCS具备多酶模拟功能,且彼此协同发挥作用。其类CAT活性分解H2O2生成O2,缓解肿瘤微环境中的缺氧状态,从而增强类GSHOx和NOX活性。GSHOx可消耗抗氧化剂GSH,NOX则阻断GSH的再生,两者协同生成H2O2,为类POD活性提供底物。持续的H2O2供应和GSH消耗共同保障·OH的持续产生,实现高效癌细胞杀伤。
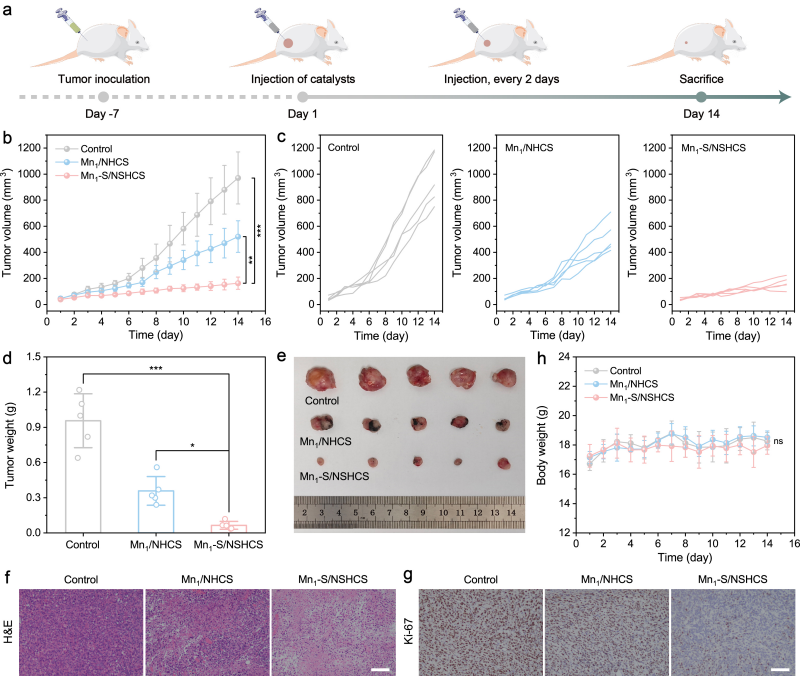
图5. 体内抗肿瘤治疗效果
Mn1‒S/NSHCS作为一种人工多酶系统,展现出优异的抗肿瘤催化治疗效果以及良好的生物安全性。
四、【总结与展望】
本研究开发了一种新型Mn1‒S/NSHCS人工酶,通过构建原子级分散的Mn–S催化对,实现高效的多酶模拟催化。实验与理论研究表明,Mn位点主要负责底物的吸附与活化,其活性受到相邻S原子的电子调控而增强;S位点则作为第二活性中心,有助于稳定反应中间体并促进产物脱附。该金属–非金属双位点的协同机制打破了传统单位点催化中吸附与脱附难以兼顾的限制,显著提升整体催化效率。凭借多种类酶活性的协同耦合,Mn1‒S/NSHCS在肿瘤催化治疗中展现出优异性能。该工作不仅揭示了催化对协同作用的关键性,也为高效人工酶的理性设计提供了新的思路。
原文详情:
Zhang, Z., Li, F., Xi, S. et al. Atomic Metal–Nonmetal Catalytic Pair Cooperatively Drives Efficient Enzyme-Mimetic Catalysis. Angew. Chem. Int. Ed., 2025. https://doi.org/10.1002/anie.202508651
本文由张哲野供稿