第一作者:李钦、兰建宏
通讯作者:顾少楠、周家东、王美玲
通讯单位:太原理工大学、北京理工大学、齐鲁工业大学
论文DOI:https://doi.org/10.1002/adfm.77151
【全文速览】
碱性阴离子交换膜水电解(AEMWE)被视为具有应用前景的绿氢制备技术之一。然而,碱性电解质中析氢反应(HER)动力学缓慢,主要受限于额外的水离解能垒。现有研究表明,引入亲氧位点可促进OH吸附,加速H生成;但随后H*在活性位点上的吸附与脱附行为往往难以兼顾,成为限制催化效率的关键因素之一。氢溢流效应被证实可在一定程度上平衡H*的吸附与脱附过程。但在传统负载型催化剂中,氢溢流通常需要克服较长的迁移路径及金属-载体界面的额外阻力,导致整体反应速率受限。如何缩短氢迁移路径、降低界面迁移势垒,成为提升碱性HER催化效率的重要科学问题。
基于上述背景,太原理工大学王美玲联合北京理工大学周家东和齐鲁工业大学顾少楠提出利用限域-热解策略,在Mo2C载体中构建铂原子岛结构,建立了一条“无界面”超短氢溢流通道。实验表征与密度泛函理论计算共同揭示,原子岛内不同配位环境的铂原子(Pt1、Pt2、Pt3)分别承担H*快速吸附、热中性迁移及H2高效脱附的阶梯式功能,从而将氢溢流路径从传统负载型催化剂的跨界面长程迁移缩短为原子尺度内的连续过程。
【研究工作亮点】
本研究在Mo2C载体中构建了铂原子岛这一介于单原子与纳米团簇之间的新型原子级结构。通过限域-热解策略,实现了铂原子在Mo2C晶格内连续取代Mo位点,形成由铂原子组成的原子岛。该结构的关键创新在于:原子岛内不同配位环境的铂原子(Pt1、Pt2、Pt3)因与Mo2C载体电子相互作用强度的差异,自发形成多功能一体化活性中心,在同一纳米尺度单元内集成了H*吸附、迁移和脱附三种互补功能。这与传统催化剂中需借助金属-载体界面或不同组分间协同工作的模式有本质区别。
本研究的核心科学贡献在于,通过原子岛结构在单组分催化体系内部实现了类似单相催化剂的原子级氢溢流效应。该设计从根本上规避了传统负载型催化剂中氢物种跨过金属-载体界面所需克服的额外能垒及长程扩散路径。实验方面,氢/氘动力学同位素效应(KIE)定量对比表明,Ptai@Mo2C@PC的KIE值显著高于Ptsa@Mo2C@PC,说明在单原子体系中氢迁移是限制整体反应速率的主要动力学瓶颈,而原子岛结构的引入使该步骤的动力学障碍得到显著缓解。理论计算则进一步揭示了两类催化剂在氢迁移路径上的本质差异:对于Ptai@Mo2C@PC,H从Mo位点解离水后,依次经Pt1(强吸附,ΔG = -0.33 eV)→ Pt2(热中性,ΔG = -0.11 eV)→ Pt3(弱吸附,ΔG = -0.03 eV)完成阶梯式溢流,决速步能垒仅为0.27 eV;相比之下,Ptsa@Mo2C@PC中孤立Pt位点缺乏相邻原子的协同接力,H需直接从强吸附位点脱附或经由载体表面长程迁移,相应能垒高达0.33 eV。上述实验与理论对比共同证实,原子岛结构通过提供连续的吸附能级梯度和超短迁移路径,有效降低了氢溢流过程中的能量障碍
【数据概览】
要点1:Pt原子岛催化剂(Ptai@Mo2C@PC)的合成路径及结构特征:
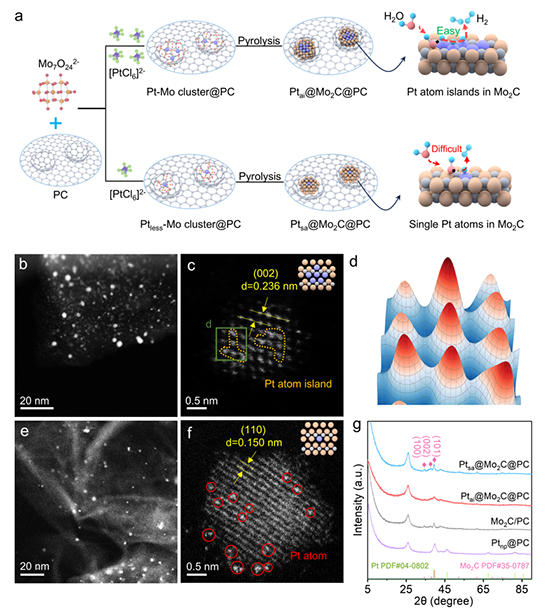
图1、Ptai@Mo2C@PC 和 Ptsa@Mo2C@PC 的合成示意图。 (b,c) Ptai@Mo2C@PC 的 HADDF-STEM 图像。 (d) 沿 (c) 中的绿色矩形截取的三维 (3D) 表面强度剖面。 (e,f) Ptsa@Mo2C@PC 的 HADDF-STEM 图像。 (g) Ptai@Mo2C@PC、Ptsa@Mo2C@PC、Mo2C/PC 和 Ptnp@PC 的 XRD 图。 Ptai@Mo2C@PC 和 Ptsa@Mo2C@PC 的示意图如图 1c、f 所示;Pt原子岛用黄色框标记,而孤立的 Pt 单原子用红色圆圈突出显示。
图1 从合成-形貌-晶相三个层面,证实了Ptai@Mo2C@PC中铂原子岛的成功构建,并揭示了其与单原子催化剂在结构上的本质差异——即原子岛内Pt-Pt相邻原子及相应的晶格应变效应,为后续探究其多功能活性中心及氢溢流机制提供了结构基础。
要点2:配位信息等的表征
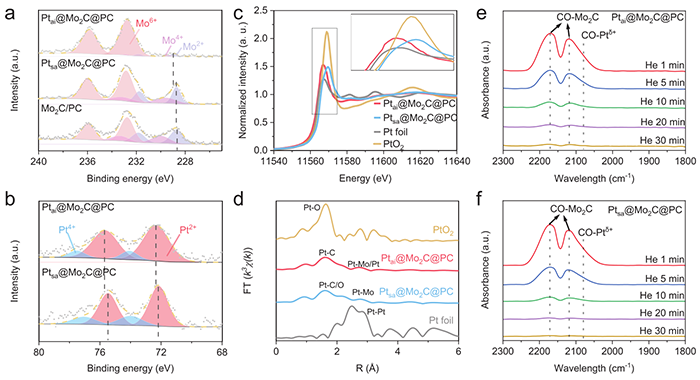
图2 (a) Ptai@Mo2C@PC、Ptsa@Mo2C@PC 和 Mo2C/PC 的 Mo 3d 的 XPS 谱。 (b) Ptai@Mo2C@PC 和 Ptsa@Mo2C@PC 的 Pt 4f 的 XPS 谱。 (c,d) Pt L3 边缘的 XANES 光谱和 FT-EXAFS 光谱。 (e,f) Ptai@Mo2C@PC 和 Ptsa@Mo2C@PC 解吸过程后吸附的 CO 的 CO-IR 光谱
图2中XPS显示Mo向Pt发生电子转移,且原子岛内Pt的结合能高于单原子体系,归因于电子在多个Pt原子间的离域化效应。EXAFS定量拟合得出Ptai中Pt–Pt配位数为5.1、Pt–Mo配位数为8.1,确认Pt取代Mo晶格位点形成原子岛。CO红外从分子探针层面排除了金属态Pt颗粒的存在。
要点3:HER性能测试:
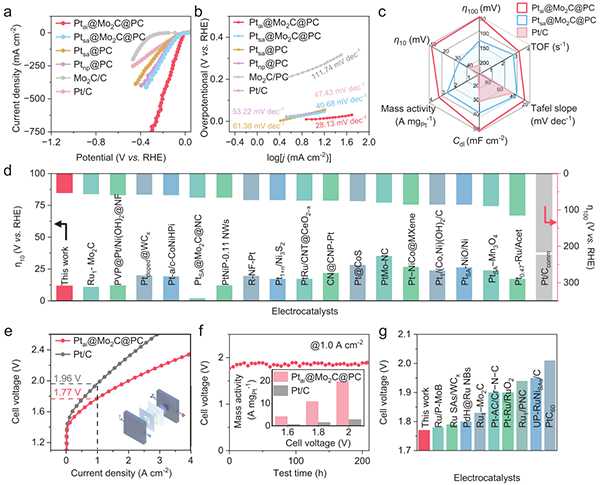
图3 Ptai@Mo2C@PC的催化性能
图3显示Ptai@Mo2C@PC在碱性HER中过电位(η10=11.8 mV,η100=52.9 mV)、Tafel斜率(28.13 mV dec-1)及质量活性(3.13 A mg Pt-1)均显著优于Ptsa@Mo2C@PC及商用Pt/C。AEMWE器件在1.0 A cm-2工业级电流密度下仅需1.77 V,稳定运行200小时无衰减,综合性能在已报道的AEMWE阴极催化剂中处于领先地位,验证了原子岛结构在实际器件中的可行性与稳定性。
要点4:内置氢溢流验证
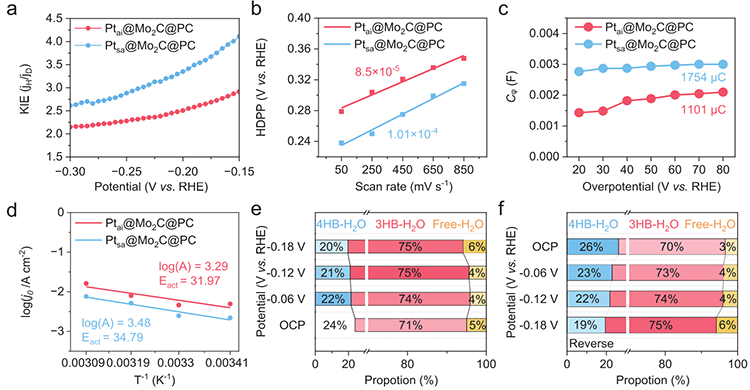
图4 氢溢流的见解
图4氢/氘动力学同位素效应(KIE)显示Ptai的KIE值显著高于Ptsa,定量表明原子岛结构使氢迁移步骤的动力学障碍得到显著缓解,而单原子体系中氢迁移是限制整体反应速率的瓶颈。CO剥离与Pt中毒实验进一步确认Pt为活性中心,原位光谱未检测到OH积累,间接支持原子岛促进H快速转移、避免活性位点被OH*毒化的机制。
要点5:内置H溢流机制:
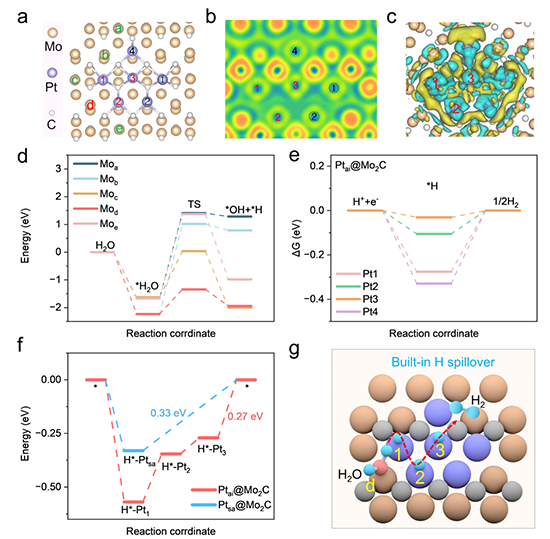
图5 理论计算探讨H溢出缓冲机制
图5理论计算验证原子岛内不同配位环境的铂原子(Pt1、Pt2、Pt3)分别承担H*快速吸附、热中性迁移及H2高效脱附的阶梯式功能。
05 总结与展望
本研究通过在Mo2C载体中构建铂原子岛结构,实现了催化剂内部多功能活性中心的集成,建立了一条内置超短氢溢流通道。实验与理论计算共同表明,该设计有效降低了氢迁移过程中的能垒,显著提升了碱性HER动力学性能。该工作为负载型催化剂中氢溢流效应的调控提供了可参考的设计思路,对碱性水电解制氢技术的进一步发展具有一定借鉴意义。
【文献信息】
Qin Li, Jianhong Lan, Xuguang Liu, Longyi Fu, Yadi Guan, Shaonan Gu, Jiadong Zhou, Meiling Wang, Advanced Functional Materials, 2026, DOI: https://doi.org/10.1002/adfm.77151.