兰州大学曾会应教授最新science:吲哚骨架单碳精准置换
[文章背景]
分子骨架编辑技术正在深刻改变有机合成与药物发现的范式。与耗时费力的从头合成不同,骨架编辑允许直接在复杂分子骨架上进行原子级插入、删除或替换,从而快速构建结构多样的化合物库。在众多具有生物活性的杂环中,吲哚骨架因其广泛存在于药物分子和天然产物中而成为骨架编辑研究的关键靶标——目前全球已有超过70种以吲哚为基础结构的药物获批上市。然而,吲哚芳香环固有的热力学稳定性使其难以被活化,传统策略往往依赖高活性的卡宾或氮烯中间体启动去芳构化,这些方法虽取得一定进展(如Studer课题组近期实现的C–N原子替换),但仍面临区域选择性控制难、副反应多以及多步操作等挑战。
兰州大学曾会应教授长期致力于绿色光化学与C–H键官能团化研究,在其前期工作中发展了系列基于可见光催化的温和转化方法。在此背景下,曾会应教授与麦吉尔大学李朝军院士合作,基于色胺衍生物中C3位乙胺侧链的内置导向作用,设计了一种全新的分子内光化学串联策略:通过[2+2]环加成/逆[2+2]开环实现吲哚骨架的去芳构化与开环,随后经光诱导脱羰基移除原C2碳原子并释放一氧化碳,最后在酸性条件下完成环化-再芳构化,一步实现吲哚C2位的单碳置换与同步官能团化。该方法条件温和(280 nm LED光照,室温,氩气氛围),无需外加金属催化剂或强酸碱,且展现出极其广泛的底物兼容性——不仅适用于芳基、烷基、酰基等各种取代基,还能成功修饰氨基酸、多肽、药物分子(如布洛芬、吉非罗齐)以及天然产物片段(如薄荷醇、表雄酮)。
尤为突出的是,该策略实现了碳-13同位素的精准引入,并应用于复杂单萜吲哚生物碱奎布拉胺的简洁全合成(仅需四步,总产率31%,此前最短路线为九步)。通过同位素标记实验、中间体捕获、光强依赖性和密度泛函理论计算,作者阐明了包括系间窜越、氧杂环丁烷开环、三氟乙酸氢键辅助降能等原子层次的详细机理。该工作将骨架编辑从依赖高活性中间体的外源策略,拓展为基于分子内光化学串联的内源精准编辑模式,为药物先导化合物后期修饰与天然产物全合成提供了高效通用的新工具。
[内容介绍]

- 创新性反应策略:基于色胺衍生物的分子内光化学串联单碳置换
研究团队利用色胺C3位乙胺侧链作为内置导向单元,将所需官能团通过酰胺键预先连接,随后在280 nm LED光照下,以三氟乙酸(TFA)为添加剂,在1,4-二氧六环中一步实现吲哚C2位的单碳精准置换。该串联反应包含四个关键步骤:酰胺负载、[2+2]光环加成/逆[2+2]开环(破坏吲哚芳香性)、光诱导脱羰基(释放CO,移除原C2碳)、酸促进的环化-再芳构化(重构吲哚骨架)。后三步在同一反应瓶中自动完成,无需分离中间体,操作简便且条件温和。
2、广泛的底物适用性与官能团兼容性
该方法可高效实现吲哚C2位的多种官能团化,包括芳基化(含供电子、吸电子、杂芳环)、烷基化(含空间位阻大的叔碳甚至季碳中心)、酰基化、氘代及¹³C同位素标记。同时,该策略成功兼容氨基酸、二肽/三肽、药物分子(布洛芬、吉非罗齐、氨甲环酸等)以及天然产物片段(薄荷醇、表雄酮)。此外,对色胺侧链N-不同保护基(N-甲基、N-苄基等)及吲哚环上不同取代位置均表现出良好至优异的产率。
3、机理验证与合成应用:严谨的机理研究与复杂天然产物全合成
通过同位素标记实验(¹³C和氘)、中间体捕获、光强依赖性实验(证实双光子过程)以及DFT计算,详细阐明了反应路径:光激发后系间窜越至三线态,羰基氧自由基对吲哚进行亲电攻击形成氧杂环丁烷,TFA的氢键作用将开环能垒从23.2 kcal/mol降至8.8 kcal/mol,随后二次光激发引发脱羰基,最后经环化-芳构化重构吲哚。在应用层面,该方法实现了复杂单萜吲哚生物碱——奎布拉胺的四步全合成(总产率31%),显著优于此前最短的九步路线,展示了该策略在复杂分子合成中的强大潜力。
[文章结论]
本研究开发了一种基于色胺衍生物的分子内光化学串联策略,实现了吲哚C2位的精准单碳置换与同步官能团化。该方法巧妙利用乙胺侧链作为内置导向单元,通过[2+2]光环加成、逆[2+2]开环、光诱导脱羰基以及环化-再芳构化四个步骤,在一锅内高效完成骨架编辑。该策略具有极其广泛的底物兼容性,可成功引入芳基、烷基、酰基、同位素标记、氨基酸、多肽以及多种药物和天然产物片段。基于详细同位素实验、中间体捕获及DFT计算,阐明了包括三线态氧杂环丁烷形成、TFA氢键降低开环能垒及双光子激发脱羰基在内的完整机理。应用层面,该方法实现了复杂单萜吲哚生物碱奎布拉胺的四步全合成(总产率31%),彰显了其在药物化学和天然产物合成中的实用价值。该工作将骨架编辑从依赖高活性中间体的外源策略拓展为基于内源光化学串联的精准编辑模式,为复杂分子的后期修饰与多样化提供了高效、温和且通用的新工具。
图文解析
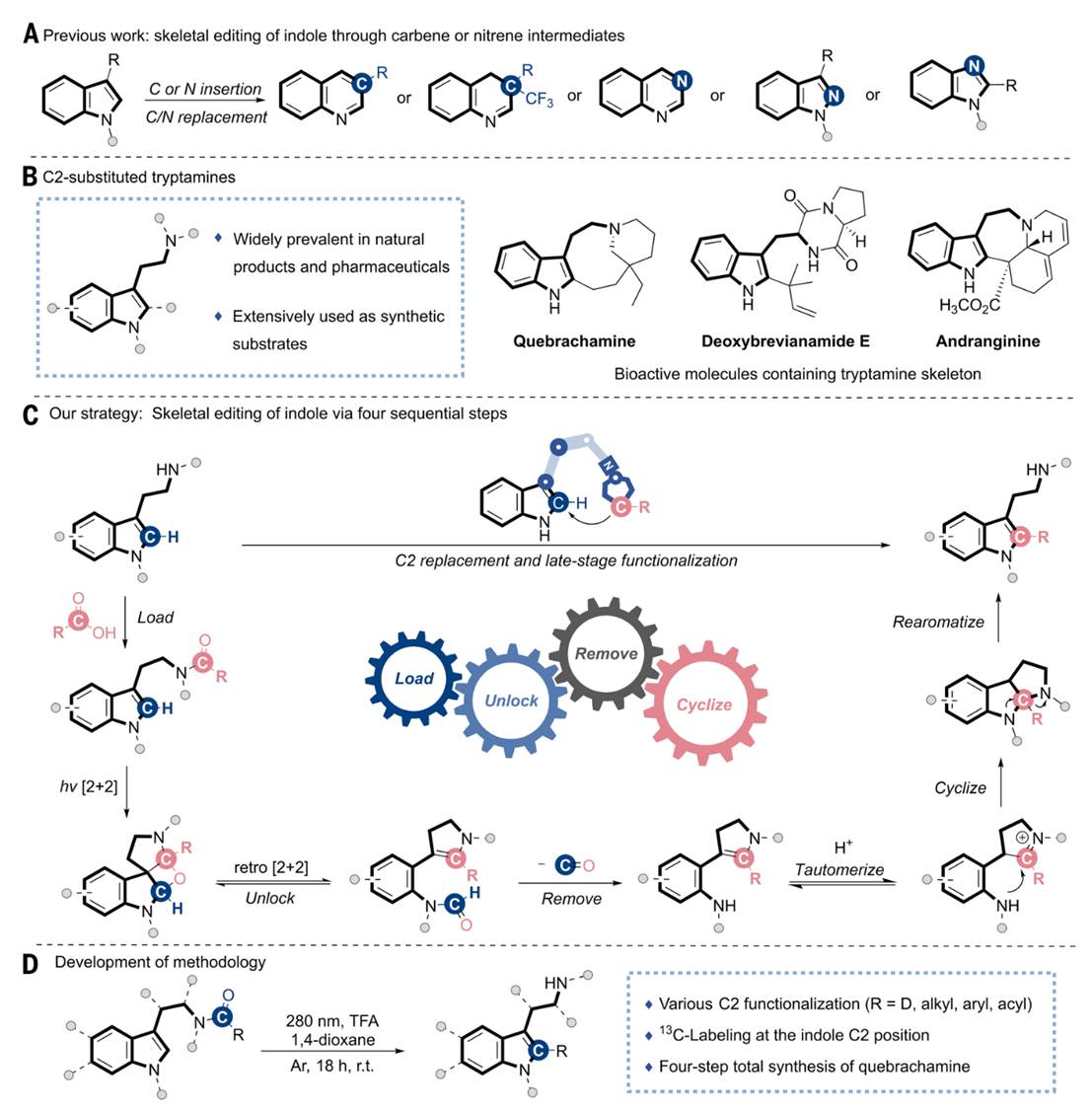
图1. 单碳置换的吲哚骨架编辑策略设计与模型反应© 2026 AAAS
图2. 吲哚C2位官能团化衍生物© 2026 AAAS
图3. N-取代基效应与底物兼容性拓展© 2026 AAAS
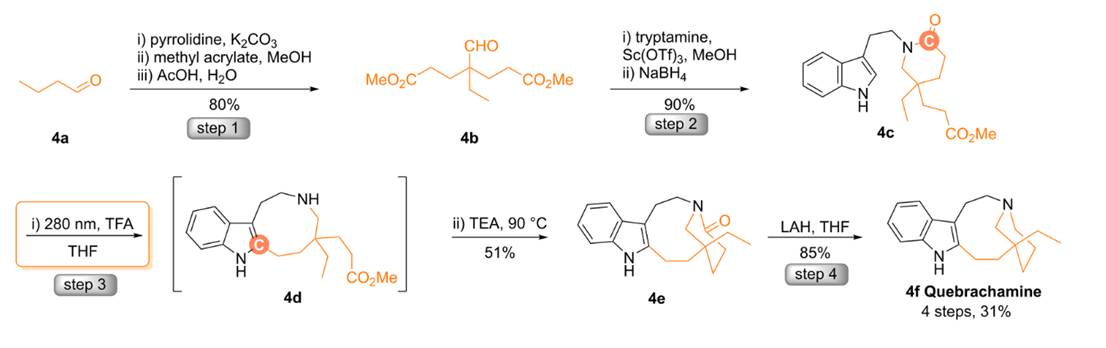
图4. 奎布拉胺的简洁全合成© 2026 AAAS
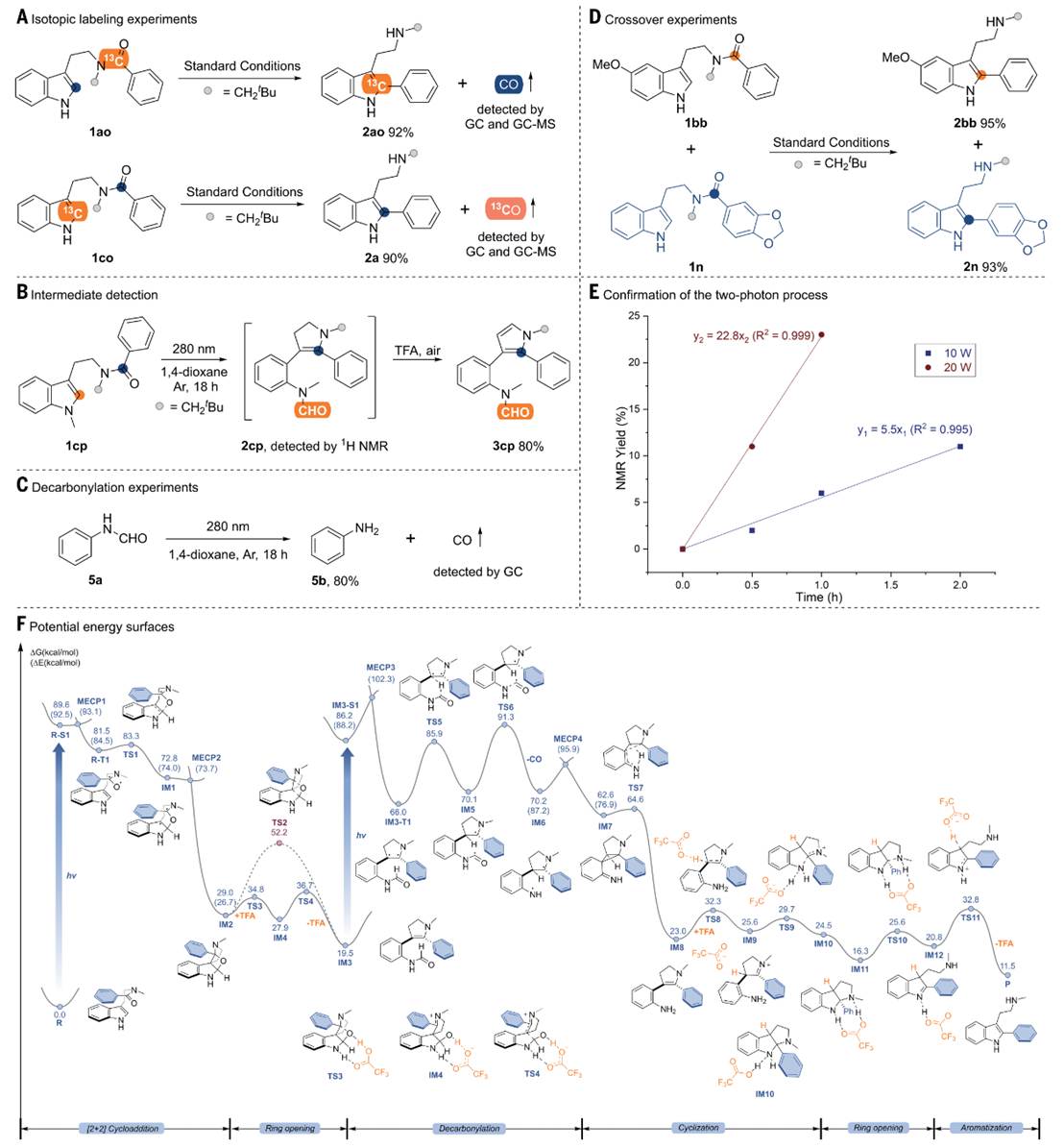
图5. 反应机理研究© 2026 AAAS
本工作开发了一种基于色胺衍生物的分子内光化学串联策略,实现了吲哚C2位的精准单碳置换与同步官能团化。该方法通过[2+2]光环加成、逆[2+2]开环、光诱导脱羰基及环化-再芳构化四步一锅转化,成功引入芳基、烷基、酰基、同位素标记、多肽及药物片段,底物兼容性极广。详细机理研究(同位素标记、DFT计算等)阐明了三线态氧杂环丁烷路径及氢键辅助降能关键步骤。应用上,仅用四步完成复杂生物碱奎布拉胺的全合成,总产率31%,显著优于此前路线。
该“内源光化学串联”策略为骨架编辑提供了全新范式,未来可拓展至其他芳香杂环(如苯并呋喃、咔唑)的原子置换反应,并发展可见光驱动的温和版本。结合流动化学与自动化平台,有望实现药物先导化合物的高通量后期修饰。此外,利用该方法精准引入同位素标记的特性,可在代谢组学与药代动力学研究中发挥独特价值。
原文详情:https://www.science.org/doi/10.1126/science.aec3587#tab-contributors